Investigadores del Centro de Investigación del Cáncer (CSIC-USAL) demuestran ‘in vivo’ el papel esencial de FOXL2 en la iniciación y desarrollo de tumor de células granulosas de adulto (AGCT)
CSIC/DICYT Un equipo del Centro de Investigación del Cáncer de Salamanca (CSIC-USAL) ha demostrado en un modelo de ratón el papel clave que cumple la mutación genética de un solo gen en la iniciación y desarrollo de los tumores de células granulosas de adulto (AGCT), un tipo de cáncer de ovario. Los resultados, que aparecen publicados en la revista Cancer Research, ayudan a comprender la naturaleza y los mecanismos moleculares del gen que codifica la proteína FOXL2, el cual está relacionado con la mayoría de estos tumores.
Cuando el óvulo se prepara para ser liberado en la ovulación, las células de la granulosa que lo rodean secretan un líquido folicular que crea una cavidad o antro. Esta masa de tejido, líquido y óvulo, recibe el nombre de folículo de Graaf. Los tumores de células de la granulosa de adulto (AGCT) son el tipo más común de tumores no epiteliales del ovario. Los AGCT ocurren con mayor frecuencia después de la menopausia y tienen un pronóstico favorable. Las recurrencias agresivas, a menudo fatales, pueden ocurrir hasta en el 50% de los casos diagnosticados. Como dichos tumores crecen lentamente, pueden reaparecer incluso 30 años después de la extirpación del tumor primario.
La mayoría de estos tumores presenta una variante en el ADN del gen que codifica la proteína FOXL2. Además, existen variantes en FOXL2 heredadas por la línea germinal que son responsables del síndrome de blefarofimosis, una enfermedad hereditaria que se caracteriza por una malformación del párpado y el desarrollo de insuficiencia ovárica prematura en muchas de las mujeres que lo padecen.
FOXL2 es una proteína que se une al ADN regulando la expresión de numerosos genes en diferentes tejidos, como el tejido mesenquimal en desarrollo que dará lugar a los párpados y a los folículos ováricos, así como en los ovarios de mujeres adultas. Su función es esencial para la diferenciación y el mantenimiento de los ovarios, así como para la represión del programa de diferenciación testicular durante el desarrollo embrionario.
La mayor parte de los tumores sólidos se caracterizan por presentar varias mutaciones en diferentes genes (oncogenes o genes supresores), que son los responsables causales del desarrollo y progresión del cáncer. Mediante la secuenciación masiva del genoma de AGCTs, se ha determinado desde hace más de una década que una de las numerosas mutaciones presentes en sus genomas se localiza en el gen FOXL2 en más del 90% de este tipo de tumores. Cabe destacar que la mutación descrita es idéntica en todos los AGCTs secuenciados y consiste en una mutación puntual en el gen FOXL2. En concreto, se produce un cambio de la base citosina a guanina, que provoca la sustitución de un solo aminoácido de los 376 que constituyen la proteína completa FOXL2, esto es, cambia el aminoácido cisteína (C) de la posición 134 por un triptófano (W), denominado C134W.
En este punto, el equipo dirigido por Alberto M. Pendás, del Centro de Investigación del Cáncer, en colaboración con Reiner Veitia, de la Universidad de París, ha investigado las mutaciones encontradas en otros genes para determinar si cooperan con la versión mutada de FOXL2 en la generación de los tumores de ovario. Se trata de constatar la causalidad de la mutación C134W. Si hay causalidad, la sola presencia de FOXL2-C134W es la única conductora del inicio y progresión de los tumores de ovario.
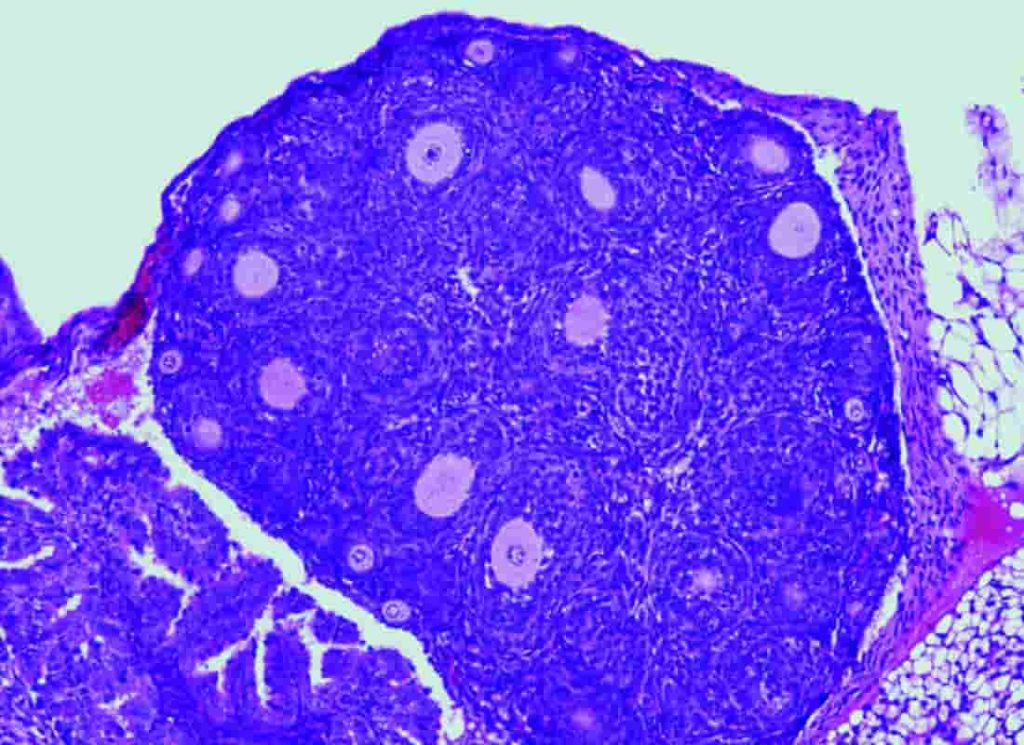
“Nuestro grupo -señala Elena Llano, de la Universidad de Salamanca- ha desarrollado por primera vez un modelo de ratón que alberga en su gen FOXL2 la variante C134W, presente en los tumores humanos, para así poder evaluar in vivo el papel desconocido de FOXL2 en la iniciación y desarrollo tumoral. Para nuestra sorpresa, estos ratones presentan la hipoplasia del párpado observada en el síndrome de blefarofimosis. Curiosamente, las hembras con dicha mutación presentan una fertilidad reducida y lo que es más relevante todas desarrollan AGCT espontáneamente. Es decir, progresan gradualmente de ovarios anormales con células de la granulosa aberrantes a ovarios con hiperplasia estromal y atipia, que finalmente dan lugar a la aparición de tumores ováricos en la totalidad de los animales antes de los 18 meses de edad. Por tanto, este proceso parece estar impulsado únicamente por la presencia de la variante FOXL2”.
Pendás resalta: “Cuando comparamos estos datos con resultados previos en AGCTs humanos observamos vías desreguladas similares. Finalmente, un análisis mutacional de los datos transcriptómicos de los AGCTs del ratón sugirió la ausencia de mutaciones conductoras adicionales además de FOXL2-C134W. Estos resultados proporcionan un claro ejemplo in vivo en el que la mutación de un solo gen desencadena el desarrollo de un tumor con profundas alteraciones en la expresión de numerosos genes cruciales para la homeostasis normal del ovario”.